knowing01 Product Information
You are working on Version 1.2 released on April 12th, 2026.
The Product Information page serves as short guide for working with our app and provides background on what is happening. We are continuously adding more information. In case your question was not answered here, just drop us a mail.
Main updates in 1.5:
-
Improved Download Center
If a dataset is re-uploaded or re-linked after an export was generated, the affected downloads are now marked as invalid rather than silently removed. Your full download history stays visible in the Download Center, so you always know which exports reflect the current state of your data foundation and which ones need to be re-run.
Short guide
The knowing01 app assists you in exploring and connecting your experimental data quickly and independently.
Getting started:
- Upload Studies, Datasets, and Hooks with a few clicks: Studies Upload, Datasets and Hooks Upload
- Set significance cutoffs: choose two cutoffs for each Dataset or Hook in Dataset details
- Explore your data and learn how your data relates to each other
- Contextualize your data and use Search One to query a single gene, protein or genomic position or Search Many to use the top 20 observations of a selected dataset or hook as your query
- Download or explore results for further analysis in Download Center or return back to the lab for follow-up experiments
About the Cellmap: The major ingredient of the knowing01 app is our proprietary Cellmap, a knowledge-graph that enables automatic translation between your data at unprecedented speed. To leverage the full information from the Cellmap, we automatically link your data to it directly after file upload.
Get in touch: If you are stuck or something is unclear, we want to know, help and do better. We would love to hear from you at any time.
Product feature guide
Studies Upload
Each study groups one or more datasets under a shared scientific context. While only minimal information is required to create a study, the more detail you provide, the better the app can support downstream analyses, grouping, and interpretation.
Study metadata:
The name will be used to represent the study throughout the entire app. We recommend choosing a concise label, for example the first author, publication year, and journal (e.g. Li et al. (2025, Nature)).
The relevance and methodological quality fields allow you to rate the applicability and scientific rigor of the study for your project. This helps prioritize studies during analysis. Rating is based on a three-tier scale: low, mid, and high, where high indicates strong relevance or excellent quality, mid indicates moderate relevance or acceptable quality, and low indicates limited relevance or methodological concerns.
Provide the original title, authors, year, journal and a link to connect the study to its primary publication. This information is displayed in the study view and can be used to trace results back to the source literature.
Use the summary field to briefly describe the study's scientific background, key findings, and relevance to your research question. This may include the disease context, patient cohort, experimental design, or key conclusions.
Figure
Upload a figure formatted as JPG, PNG, or similar image format. Include the figure legend to provide context and support interpretation.
Study numbers
The study numbers table captures quantitative information about participants, samples, assays, control groups, disease groups, and cell/nuclei counts. To enter study numbers:
- Specify each assay used in the study
- Enter the number of participants involved in that assay
- Enter the final sample number for the assay
- Select a condition (e.g., disease/control samples or cell/nuclei count)
- Specify the exact number within that condition
- Optionally, add details such as the specific disease (e.g., Breast cancer)
Study tags
Add study tags by typing and pressing Enter. Use tags to label studies according to disease area, species (e.g., human, mouse, rat), technology, or research theme. Tags facilitate grouping across studies in later analyses and should be true for all downstream linked datasets.
Comments
Use the comments field for internal notes or curation remarks that do not belong in the summary.
Datasets
Existing datasets can be linked to a study under Datasets. This connection allows the app to provide study-level context for all associated data.
Studies View
Once you complete the study creation, it becomes available to all your project members in the knowing01 app. In the left-hand navigation of the Datacenter, you see all your studies and linked datasets or hooks with their respective processing status. Clicking on a study displays its details. The Studies view presents the key metadata you entered, including name, authors, year, journal, summary, uploaded figure, study numbers table, and linked datasets. Studies can be marked as favourites and sorted.
Datasets and Hooks Upload
Each file uploaded is transformed into one dataset or hook. Datasets contain your own experimental data, while Hooks are personally created lists or results from the Contextualize and Explore results. While only minimal information is required for the transformation, the more information you provide, the better the app can leverage the potential of your data.
File and Information
You can upload any text file in tabular format. Common file endings of such files are .CSV, .TSV, or .TXT. We automatically detect headers in your file and recognize common field delimiters, such as tabs or commas.
Biotype
The biotype guides the linking of the dataset to our Cellmap. The app supports three top-level biotypes: DNA, * RNA*, and Protein. Within each, you select a specific second-level biotype such as Variant, Genomic Range, Gene, Transcript, Protein, or Gene Symbol. Read more on the current scope of our Cellmap to learn about how the biotype information influences your analysis results.
- For the top-level biotype DNA and second-level biotypes Variant and Genomic Range, you need to specify the genome assembly version. If you are unsure, please request this information from your data provider. Commonly used assembly versions of human genomes are provided.
- For the top-level biotype RNA and second-level biotype Gene Symbol, you need to specify the organism. Especially for rodents, gene names may be identical, so the taxonomy cannot be directly inferred from data alone.
Column roles
We ask you to assign a role to the columns of your file. Information from all assigned columns will be displayed during analysis, so we recommend assigning roles to as many columns as possible.
- Assign columns containing significance and effect size (e.g., fold changes) values to differentiate between relevant (e.g., highly significant) and irrelevant (e.g., non-significant) observations after the dataset is fully integrated. Specify the direction of effect size in the metadata below.
- Assign the "Link Identifier" role to the column containing the molecular/reference database identifier (e.g., UniProt ID or accession, ENSEMBL gene ID, Gene Symbol, dbSNP ID, etc.) (for further details see Processing). This is essential to ensure meaningful linking of the observations to the Cellmap.
- Additional column roles that are available:
- Score: A score is a positive value where higher numbers imply greater relevance. When a score is used, the dataset or hook may be classified into stringent and lax cutoffs by using the top 10% and top 20% percentiles of this column. Please contact support for assistance with this handling.
- Data Slice: It may occur that the uploaded file contains several types of comparisons in a long format, e.g., all marker genes per cell type or pathway names of a gene-to-pathway master table. Use the Data Slice role for these typically categorical columns.
- For Single Cell Datasets: The PC1 and PC2 columns are often available. These refer to the fraction of cells in which the feature is found within the cluster and outside the cluster, respectively.
- Use our custom roles 1–5 for columns that are of relevance during your analysis.
Note that in every export you will receive the full file information, including unassigned columns. We are not losing your data, but try to reduce complexity with and for you during exploration.
Dataset and Hook metadata
The dataset title will be used to represent the dataset or hook throughout the entire app. We recommend providing a crisp and concise title, including assay type, biosample, and result type (e.g., PWAS - T2D - Plasma Proteins or ATAC peaks - T2D islets).
Parent Study you can connect the dataset or hook to an existing study. This connection allows the app to provide study-level context for the data.
Use the description to provide a brief summary of the data in the file, including measurement methods or technologies.
Provide the effect size definition to clarify what the effect size values represent in your dataset based on the effect size column assigned above. It is important to specify the direction: whether higher values (e.g. fold changes) indicate increased risk in cases or controls, as this can vary across different studies.
Add analysis model information to describe the underlying sample sizes, quality control filters, bioinformatic pre-processing, and statistical methods used.
Add dataset tags by typing and pressing Enter. Use tags to label datasets according to experiment type, species, technology, or indication group. This will facilitate dataset grouping in later analyses. Tags should be specific to the dataset and not overlapping with study tags.
Add the data source (e.g., Supplementary Table 1) to trace back to the original data location.
Use the comments field for internal notes or curation remarks that do not belong in the description.
Datasets and Hooks Views
Once upload is finished and the app processes the file and transforms it into a dataset or hook. Afterwards it is also available to all your project members in the knowing01 app. In the left hand navigation of the Data center, you see all your datasets or hooks with their respective processing status. Clicking on an integrated dataset, its details are displayed.
The top of the detail view covers the key metadata captured during upload, such as title, description, original filename, as well as the selected biotype and analysis model. Furthermore, it contains basic statistics on the number of observations extracted from the file and how many of those observations were linked into the Cellmap and can thus be translated for analyses across biological data types.
If assigned, the significance values are used to extract relevant observations for explore analyses (Read more on significance cutoffs. You choose a stringent and a lax cutoff from a list of commonly accepted significance cutoffs. The number of observations considered for each cutoff are there to help you choose a meaningful cutoff. The choices here directly impact which observations are taken into account for analyses throughout the application, i.e., in the Explore view.
The assigned column role section allows you to review the roles provided during upload, which determines how the dataset is used in the app. In case, any column role needs to be updated, please use the edited mode.
Contextualize
Choose between two options: Search One and Search Many.
Search One: Search for a human, murine, or rat gene, protein or genomic position and retrieve all observations across all your available datasets. The app auto-translates the search term to all matching biotypes.
Search Many: Search for a Dataset and automatically use the top 20 observations as query for the human, murine, or rat gene, protein, or genomic position observations that are to be found across all your available datasets. The app auto-translates the search term to all matching biotypes.
How it works: The query will first be linked to all matching Cellmap nodes. Especially for Gene Symbols this might result in multiple hits. Second, the Cellmap hits are auto-translated before, finally, being searched for across your available datasets. Search results will show all matching Cellmap nodes, which may be also de-selected by you in order to filter the search results.
All results are consolidated in a single, comparative view, allowing for intuitive comparisons of data distribution and quality across datasets at a glance. The view is interactive, enabling users to filter results by adjusting rows and columns, as well as modifying cutoff criteria to explore the data in greater detail.
The search results are summarized in a visualization that provides a detailed, comparative view of observational data across matched datasets within the same group. Each dot represents the number of observations, with dot sizes indicating relative quantity. The dot pie charts further break down these observations, showing the proportion that meets specific cutoff criteria for each dataset group and match. All results are now consolidated in a single, comparative view, allowing for intuitive comparisons of data distribution and quality across datasets at a glance.
The Search results are all observations that were found in your available datasets. You may subset the results further by whether they match to each dataset's stringent or lax cutoff criteria.
Our search feature includes a convenient export button. When you request an export, the resulting file is generated and made available in the download center. You can easily access and retrieve your data from there, ensuring you have all the information you need for further analysis or reporting.
This feature was released in beta version. Please provide feedback for improved visualizations and usability.
Explore data
In the left panel of the Explore data view, datasets can be searched and iteratively combined with the aim to identify joint or unique components. Only after adding a dataset, the dataset combination is computed. In the main panel, the explore results are shown or previewed in case of large results. Components are generally ranked by significance.
If observations are comparable from a biological perspective, we group them into so called Cellmap components (find below all details on components). Amongst others, we are composing homology components ( human/mouse/rat), variant/SNP proxy components (using Linkage disequilibrium information) as well as genomic range/variant components.
Note that Explore translations are directly bound to the current version of the Cellmap. For other datasets, Explore is intersecting/subtracting observations solely based on values obtained from the file. The translation functionality is continuously being extended. If you have a specific analysis need, simply reach out to us, and help us prioritize!
Basic analysis flow
-
An analysis is started by selecting the first dataset. The select field lets you search through shortname and description of the datasets. After selecting the dataset the choice between the customizable significance cutoff scenarios are left, before you can add the dataset to the analysis. Per cutoff, the number of observations that adhere to the cutoff are shown, to guide the parameter choice.
-
The result preview displays the overview of the initial dataset selected, showing the top 500 components by significance of the respective observations, provided that the significance column has been tagged for the dataset during upload
-
Before adding the next dataset to the analysis, you need to choose between two set operations:
 INTERSECT: Will return in the preview those components that can be found in both,
the current result of the
already combined data, and next selected dataset selected.
INTERSECT: Will return in the preview those components that can be found in both,
the current result of the
already combined data, and next selected dataset selected. SUBTRACT: Will return only those components in the current state of the analysis,
that are not found the next
dataset for consideration.
SUBTRACT: Will return only those components in the current state of the analysis,
that are not found the next
dataset for consideration.
-
After choosing the set operation, the next dataset can be added to the analysis, and so on.
Export preview
The result preview intends to provide an overview of the most relevant explore results. Ranked by significance, the preview components are of highest significance (e.g. lowest p-value). The backend server stops collecting more explore results once 500 components have been assembled.
Depending on the connectedness of observations in our Cellmap, components may be comprised of more observations than query datasets. The component size histogram will give an overview of the component sizes of the preview (Figure 1).
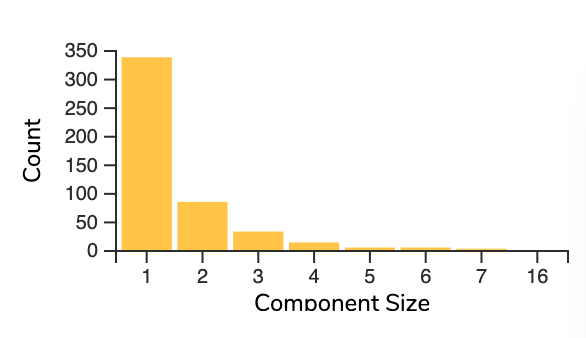
Figure 1. The number of components in the explore preview are shown according to the component size itself.
You may filter the components by concordance of effect size: up- or down-regulated or mixed regulation. To obtain the full results, please request an export and download them in the Download Center.
Export results
To receive an export of any of a specific Explore analysis step, simply use the "Export results" button. Choose your desired format the dialogue.
For Explore analyses with datasets of exactly two variant biotypes, a button is shown that allows you to trigger a specific export, a so called "Variant Proxy Export". You are prompted to indicate the target dataset, i.e., the dataset that shall be exported using the other (the "link" variant) file's information. There is a choice between to r2 cutoff values to be considered when generating the export.
All export requests are generated automatically and will be available through the Download Center.
Download Center
All your export requests are sent to the Download Center. Large-file requests may take some time to process. Once finished, the files are ready for download.
Background
File processing
After uploading the file to your project's repository at knowing^01, the file will be processed in two steps before becoming available as dataset. The progress is shown in the data status field.
- During import, the file is processed on the basis of the provided biotype and tags and the significance cutoffs. In this step we may run into errors when the file format changes in between or unforeseen field values are encountered.
- Special Case: During import, the Significance and Adjusted Significance values are analyzed for putative transformations. In some cases, the uploaded files contain values that were log10 transformed. Whenever we detect values out of (0,1) range, we assume -log10 or log10 transformed values. We automatically perform back-transformation, so you are able to work with the cutoff filters as intended.
- During linking, we are establishing the connections to the Cellmap.
The information on biotype plus the tagged columns is used to link the entries/rows of a file (lets call them * observations*) to the knowing^01 universe. Imagine that each observation is going to be plugged into our universe according to your tagged information (Figure 2). So, as of today, the more and comprehensive your tags are, the better we can work with the data. We are continuously learning in order to automate this in the future. Check out this figure to better understand the idea of tagging the correct column with what we call "Link Identifier".
The (adjusted) significance tags are used to extract relevant observations for all explore analyses, focusing the results displayed. Only with significance tags, we are able to focus the result presentation to the most relevant observations. You may choose amongst different significance cutoff options for your Explore analysis in the Dataset details view. During the initial upload, the extraction occurs according to the following approach:
- If "Adjusted Significance" was tagged, we extract observations with "Adjusted Significance" < 0.05.
- Else, if "Significance" was tagged, we extract observations with "Significance" values < 0.01.
- Else, if neither of the fields are tagged, all observations are considered to be relevant for all explore analyses.
Please note: Files are processed sequentially. An internal queuing system processes files one after another, in case several files are uploaded to the application in a short time. Processing may take longer for files with millions of observations.

Figure 2. Minimally required information per file for explore analysis. Left: The (adjusted) significance column will be used to split the data into a relevant/significant part and a non-relevant/non-regulated/unchanged part. Right: The link identifier information will be plugged into the knowing01universe, so any connections between different (bio)types of data can be made automatically.
Cellmap
Current Version: 6.0
During explore analyses, we auto-translate your data according to the current scope of our Cellmap. These translations allow us to compare data across biotypes and return observations that are related across the data sets.
The scope of the current Cellmap version determines the links between datasets that we are establishing. We may differentiate between the biotypes and their links (aka. translations between the biotypes).
Biotypes
Making use of public reference databases we are assembling our Cellmap to the best of use for your analysis. Version 006 scope encompasses the following information, grouped by biotype.
- Proteins are here defined as those proteins and their isoforms defined by the commonly used protein-centric database UniProtKB. The UniprotKB is a collection of verified proteins (SwissProt) as well as automatically translated mRNA sequences from EMBL, called trEMBL. For example, the protein of the human gene TBC1D9B has the UniProtKB identifier " Q66K14".
- Gene Symbols are the typically short gene names we know, like TP53, the gene of the famous tumor protein maker p53. There are world-wide efforts to harmonize the gene symbols used for any gene resource. It is important to note that gene symbols are subject to many changes, so the synonyms are frequently in use, without knowing. Another famous example is the key transcription factor and proto-oncogene PU.1 that is referred to SPI1 nowadays. In short, the exact Gene Symbols are quite dependent on the month and year the dataset were generated.
- Genes are here defined as genes as defined by major gene reference databases, such as ENSEMBL. They are best referred to by their (rather cryptic) identifier e.g. ENSG00000066336. From a bioinformatic point of view, these identifiers are unique, easy to find, and work with in a non-ambiguous manner.
- Variants are defined as 1 base pair (bp) positions on a chromosome of a reference genome.
- Genomic Ranges are defined as ranges (with start and stop position) on a chromosome of a reference genome.
Translations
The following translations between individual bio-(sub)types are being made. Figure 3 illustrates the Cellmap knowledge graph in a graph model.
- Proteins are linked to ENSEMBL genes, while isoforms are linked to ENSEMBL transcripts, according to the data provided by UniProtKB.
- Gene Symbols and all synonyms are obtained from genome organisation consortia. More specifically HGNC for human, MGI for mouse and RGC/RGNC for rat.
- Gene files with ENSEMBL identifier that corresponds to human, murine (mouse) or rat. This homology information is obtained from ENSEMBL database and directly reflects efforts of the COMPARA initiative. ENSEMBL Version 101 is used. Links between Gene Symbol and Genes are included, too.
- Variant files with Linkage disequilibrium information, with r>0.2, GRCh37, 1000Genomes, Phase 3 V5, and European ancestry. Note that assembly number GRCh37 is identical to hg19.
- Genomic Ranges are linking to each other whenever there exists an overlap of 1 base. For examples a range from chr1: 1-10 overlaps the range chr1:10-900. The ranges on assembly hg19 are readily linked to variants.

Figure 3. The Cellmap 6.0 Depicted in a graph model, the Cellmap is knowledgable about: Gene Symbols with their synonyms from human/mouse/rat and species-specific gene links. Genes and their homologs from again the three species; Proteins, their isoforms link to transcripts and genes, depending on their relationships known today. LD information only available for hg19 human genomes. Genomic ranges are connected to variants if variant co-localized in the range.
Components
When selecting one, two, or more datasets during explore analysis, we are grouping the observations of the dataset(s) into so called "components", whenever they are comparable biologically speaking ( c.f. Scope of our Cellmap).
We are working on biologically relevant component names. In the meantime, we are simply giving random names to the components for the sake of identifiabilty.
Example: Variant Components
Let us look into the example of variants that are co-inherited, so are in a so called linkage disequilibrium (Figure 4). Imagine we selected dataset "A" in explore, that contains genome-wide significant hits of a recent Genome-wide Analysis Study (GWAS). Amongst the most significant common variants, or Single Nucleotide Variants (SNPs), there are six with positions on chromosome 21 from 34,593,673 - 34,626,654. When consulting a linkage disequilibrium (LD) resource, we may notice that the six SNPs are in high LD, as measured using the r2 genotype correlation value. In the results of the explore analysis, hence, is a component with those 6 SNPs. You may easily pinpoint the number of independent loci in the full dataset GWAS "A".

Figure 4. Variant components explained. ( Left) Dataset "A" has six significant variants on chromosome 21. (* Middle*) All variants are in high LD to each other. ( Right) When selecting dataset "A" in explore, we group all variants in high LD into a so-called "component". This approach works analogously for combining two or more dataset: one component is returned in explore, when intersecting two datasets and each dataset "hits" the LD block with one or more variants.
Example: Homology
Let us investigate the component as explore results in the situation of homology. Imagine we have been analysis disease tissue expression with a blood group effect. To that end we analyzed gene expression in three species, namely human, mouse, and rat (Figure 5). We aim to know the overlap of the three datasets with few clicks. In our example the alpha 1-3-N-acetylgalactosaminyltransferase and alpha 1-3-galactosyltransferase gene is significantly regulated in all three species. From our Cellmap we know about the homology although genes are named differently in the different species.
Let us select the three datasets in the explore module and ask to intersect them. As one result hit, the ABO/Abo component is returned, finding and grouping all homologous genes into one group of biologically comparable observations.

Figure 5. Homology components explained. Imagine three datasets (called "human", "mouse" and "rat" ) are selected to be intersected, wishing for homologous genes to be captured as hit. (Left) Imagine one or more observations of this gene are found to be significant in all three datasets, yet their ENSEMBL gene identifier and their symbol varies. (* Middle*) From the Cellmap, we have information that all five human/mouse/rat genes are pairwise homologs. (Right) As a result of the intersect operation one component with all five genes are found.
Version history
In the following section, we share the main changes of the different versions of both, the knowing^01 SaaS application and the underlying knowledge graph, the Cellmap.
Application
Main updates in V1.4:
-
New Feature: Filtered Contextualize Export
You can now export exactly what you see. After narrowing Contextualize results by query match, dataset group or observation cutoff, use the export button to download only the filtered selection (not the full result). This keeps your output files focused on the observations relevant to your current question and reduces the cleanup work downstream.
Main updates in V1.3:
-
New Feature: Datacenter Dashboard
The Datacenter Dashboard provides a structured overview of your studies, datasets and hooks. Key metrics, status indicators and tag-based breakdowns are consolidated in a single view, giving you immediate insight into the state and composition of your data foundation.
Main updates in V1.2:
-
New Feature: Studies
You can now add studies and link them directly to available datasets in your data center. The Studies section is accessible alongside the existing Dataset and Hook views.
-
New Visualization: Studies View
The Studies View provides a structured summary of individual publications, consolidating key findings, a primary figure, and study numbers in a single view. Each study can be ranked by relevance and methodological quality and displays a overview of its linked datasets.
Main updates in V0.17:
-
New Feature Update: Contextualize
Now, in addition to searching one query at a time, you can enrich the top 20 observations of each dataset with contextual information—regardless of dataset biotype.
-
New Visualization
The Contextualize feature enables multi-query searches and summarizes results in a comparative view. This top-level overview displays search matches, the number of matching observations per dataset group, and details on cutoff types.
Main updates in V0.16:
- The latest feature update, Search, now includes an export option. The search results in the app provide a preview for molecules with large amounts of evidence. The export option contains the full scope.
- Datasets may now contain sets of meta tags that help users with numerous datasets introduce an additional layer of clarity alongside the dataset title. Meta tags can be added during upload and separated by a semicolon ";" if multiple tags are used.
- We introduced new column tags to facilitate more precise data descriptions that are later required for improved analysis: "Score" for classifying datasets based on percentile rankings, "PC1 and PC2" for mapping feature distribution in single cell datasets, and "Data Slice" for tagging categorical columns in datasets with multiple types of comparisons. These tags are intended to provide users with enhanced capabilities for organizing and analyzing their data effectively.
Main updates in V0.15:
- New analysis feature Search. With search, you are able to query your data across all datasets that are linked to the Cellmap. The search term will matched to its Cellmap node(s), e.g. a Gene Symbol, before being translated to all possible biotypes. Resulting hits are dataset observations reachable through the Cellmap links to your search term.
- Explore gets a summary plot for component sizes. Read more in Explore Section.
Main updates in V0.14:
- New Cellmap Version 6.0. With the new cellmap version, the auto-translation mechanism will translate in longer-range depths: like from Protein to hg38 Variant and Genomic Range information, from Human Protein to Mouse/Rat Gene Symbols. Of note, the hg19 genome assembly information is not yet coupled to the remaining biotypes.
- The Download Center was added beside Explore data Feature and the Data Center. Export requests ( explore and variant proxy exporter) become ready for download once generated. Please note, that knowing01 Support team is running our quality checks subsequently, and update the request status to "approved" after successful checks. The request status shows the current status of your request.
Main updates in V0.13.1:
- Major backend update regarding user data import and linking, which required an reimport the entire user data (starting 13. Dec. 2021). This was an essential step to take in terms of scaling and being able to process even more data in short amounts if time. Processing tasks are now fed through a queueing system also improving on data sanity checks.
- Import errors are getting fed back from the queueing system to the user, for improved user guidance. Additionally this information improves our support response times that are related to data upload and (failed) processing.
- Fixing variant-range data integrity with additional quality control steps, finally bringing full capacity of a correct and comprehensive link set to the app.
- Patch 0.13.1. further refined and scaled the queueing system.
Main updates in V0.12:
- Introducing Cellmap V004 introducing full potential of all possible variant - genomic range links. This mainly affects human genome assembly GRCh38/hg38
- Patch 0.12.1 fixes an overlap error that occurred before when combining large variant/genomic range datasets.
Main updates in V0.11:
- Reworked explore analysis algorithm on the backend to increase speed as well as expand overlap hits for sparsely connected components. Especially for huge variant files, with hundreds of variants per component, V0.11 eases the search burden.
- Small bug fix on Genomic Range linker to Cellmap. All Genomic Range files have been relinked.
Main updates in V0.10:
- Introducing proteins to the app, thus releasing Cellmap V003 and protein-gene components in Explore. Uniprot protein and isoform identifier (IDs) and accessions are used to link to proteins in the Cellmap, which are in turn connected to genes and transcripts from ENSEMBL.
- Updating component view to lower the data burden for the browser. Use load more and collapse buttons to show more hits if required.
Main updates in V0.9:
- Introducing genomic ranges to the app, as part of the Cellmap V002 release and range components in Explore. Data with genomic range data, for example BED files, may be incorporated now. Links to human hg19 variants were added, too. Note that integration of genomic ranges may take longer to integrate.
- To guide the cutoff choice in the explore view, the number of observations that adhere to the respective criterion are shown. Note that, you are may adapt the thresholds for both cutoffs in the data center view of each dataset.
Main updates in V0.8:
- Redesign of the dataset selection flow in Explore.
- Improved Explore analysis with respect to speed and information content: In shorter time, you will additionally learn if the Cellmap was employed for building a component – or not.
- Export requests are handled directly in the app. Although we like getting emails from you, the new way reduces complexity for both of us.
- Information on Cellmap linkage status of each dataset is provided in the Data center.
Main updates in V0.6:
- Every explore result is now available as export upon request. Find a export request button above every result preview.
- We are introducing two significance cutoffs, with the names stringent(default) and lax. You may now choose separate cutoff values for dataset in the data center.
Main updates in V0.5:
- Release of Cellmap Version 001. The first release encompasses: human/mouse/rat homology for ENSEMBL genes according to COMPARA consortium, gene symbols with primary and synonyms for human/mouse/rat, gene symbol to gene links, as well as variants with their linkage disequilibrium (LD) from EUR ancestry for human genome assembly hg19.
- We are introducing components, the concept to group biologically comparable observations across datasets, like grouping observations with current gene symbols and all their synonyms.
Main updates in V0.2:
- Explore with translations: When comparing data in Explore, variant datasets are translated using linkage disequilibrium as well as gene datasets from human, mouse, and rat.
- Progress information during processing: The two-step process of after uploading data to the app, importing, and linking to the Cellmap, are now made visible with progress information.
Cellmap
- V005 is an optimized version of its content-wise equivalent V004. Few special cases are now included. For example only one rat Gene Symbol actually matches two ENSEMBL Genes instead of one.
- For V004 all possible overlaps between variants and genomic ranges were established, mainly introducing overlaps also on human GRCh38/hg38 assembly.
- For V003 the protein biotype was added, modelled by proteins and their known isoforms.
- For V002 Genomic Ranges were added.
- V001 includes (ENSEMBL) Genes, Gene Homology and Gene Symbols from human, mouse, and rat as well as variants and their LD information for hg19 human genome version.